Man lädt die Rohdaten des NGS Tests nicht selber zu Yfull hoch, sondern gibt an, wo diese runtergeladen werden können. Den link hierfür bekommt man beim Anbieter, bei dem man seinen Test gemacht hat. (FTDNA, YSEQ, FGC). Firmen wie Dante Labs bieten solch einen link für Yfull nicht direkt an, so dass man die benötigte Datei über eine Cloud zugänglich machen muss.
Diese Anleitung ist einfach gehalten und für jene gedacht, welche selber gar keine Erfahrung haben, aber trotzdem Ihre Daten an YFull übertragen wollen. Deswegen werden nur die einfachen Methoden erklärt. Die „schwierigeren“ Methoden werden nur erwähnt und nicht detailliert erklärt.
1.0 WGS von Dante Labs
Grundsätzlich wird empfohlen, für den Y-Baum von YFull die Rohdaten als BAM Datei mit hg38 zu verwendet. Es ist aber auch Möglich die Analyse mit einer BAM hg19 zu starten und später ein Upgrade auf hg38 zu machen.
Ladet Eure Daten von Dante Labs runter und speichert diese lokal ab.
1.1 Einreichen der BAM mit hg19
1.1.1 Hochladen der gesamten Datei.
Die gesamte Datei ist sehr groß, so dass diese Variante nur im Ausnahmefall angewendet werden sollte. Außerdem enthält diese Datei die gesamte autosomale DNA.
- Die Dateien mit den Endungen BAM und BAI auf eine Cloud hochladen.
- Den Ordner Freigeben und die URL an YFull übermitteln.
Die Datei mit der Endung BAI ist eine Archivdatei. Mit Hilfe dieser muss YFull nicht die gesamte BAM runterladen, sondern ist in der Lage nur einen ausgewählten Bereich (Y-Chr) runterzuladen.
1.1.2 Hochladen der Y-Chr Datei.
Man kann aber im Vorfeld nur die relevanten Bereiche aus der BAM extrahieren. Dies geschieht auf Linux mit dem Programm Samtools. User ohne Erfahrungen in Linux und Samtools können ein Tool Namens WGS-Extract nutzen, welchen auf MAC und Windows läuft und im Hintergrund mit Samtools arbeitet. Dieses Programm ist deswegen sehr groß (4,5 GB), da es die Referenzdateien für hg19 und hg38 beinhaltet. Das Programm erkennt das verwendete Referenzgenom und funktioniert sowohl mit BAM hg19, als auch mit BAM hg38.
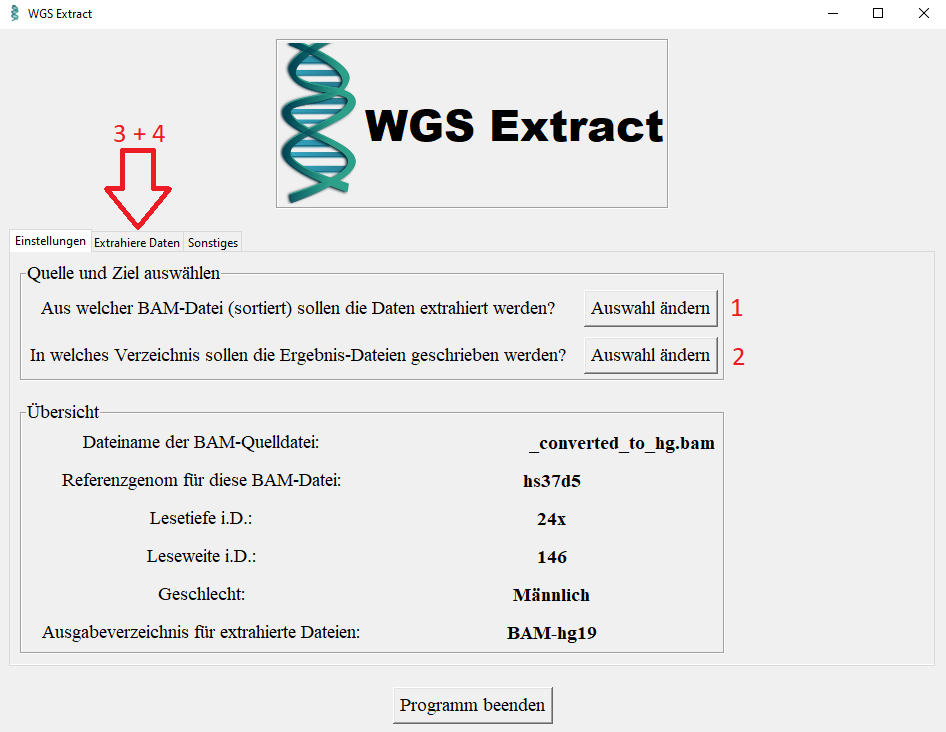

- WGS Extract hier runterladen und entpacken.
- Programm starten mit Windows_START.bat
- Sprache wählen Deutsch / Englisch
- BAM Datei wählen (1)
- Ausgabeverzeichnis wählen (2)
- BAM-Datei generieren, welche sowohl Y- als auch mtDNA enthält (3)
- Man kann auch aus einer WGS-BAM mit hg19 Dateien in verschiedenen autosomalen Formaten generieren. (4) Diese Dateien können zum Hochladen zu Gedmatch verwendet werden.
- Die Datei #######_only_chrY_and_chrM.bam auf eine Cloud hochladen.
- Den Ordner Freigeben und die URL an YFull übermitteln.
1.2 Einreichen der BAM mit hg38
Das Bevorzugte Referenzgenom für YFull ist hg38. Dieses kann man entweder selber erstellen, oder sich erstellen lassen.
1.2.1 BAM hg38 Von YSEQ erstellen lassen
Die Einfachste Methode ist, sich die Datei von YSEQ anfertigen zu lassen.
- Das Produkt von YSEQ nennt sich FASTQ Mapping to hg38.
- Es gibt vier Methoden, wie man YSEQ die Rohdaten im Format FASTQ zukommen lassen kann, damit diese in eine BAM hg38 umgewandelt werden. Überlege, welche Methode von „To transfer the large sequencing data files you have several options:“, für Dich am besten geeignet ist.
Besonders dann, wenn man selber eine langsame Internetverbindung hat, empfiehlt es sich Punkt 4) zu wählen, damit YSEQ die Rohdaten selbst von Dante Labs runterlädt. - Bestelle das FASTQ Mapping und gebe an, wie die FASTQ an YSEQ übermittelt werden sollen.
- Es kann nun einige Tage bis Wochen dauern, bis Deine Dateien fertig sind.
- Lade Die Dateien runter
- Die Dateien mit den Endungen _hg38_chrY.bam und _hg38_chrM.bam auf eine Cloud hochladen.
- Den Ordner Freigeben und die URL an YFull übermitteln.
1.2.2 BAM hg38 mit usegalaxy.eu selber erzeugen
Man kann die BAM hg38 auch selber aus den FASTQ Dateien erstellen. Das geht auf dem eigenen Rechner, aber auch auf einem ausgelagerten Rechner von usegalaxy.
1.) Man registriert sich auf https://usegalaxy.eu/ einen Zugang und loggt sich ein.
Bitte wirklich usegalaxy.eu und nicht usegalaxy.org verwenden, da Letzteres
strengere Limits einsetzt, welche die Generierung der BAM-Datei abbrechen
lassen.
Usegalaxy.eu wird von europäischen Universitäten betrieben.
2.) Jetzt muss man in den Rohdaten schauen, wieviele
FASTQ-Dateien man hat – die erkennt man daran, dass ihr Name jeweils auf .fq.gz
endet.
Falls es sich insgesamt um genau zwei FASTQ-Dateien handelt, dann ist in diesem
Schritt nichts mehr zu tun, sie liegen schon im richtigen Format vor – man kann
dann mit Schritt 3 fortfahren.
Falls es mehr als zwei Dateien sind, muss man diese erst zu zwei Dateien
zusammensetzen.
D.h. alle Dateien, deren Dateinamen auf _1 enden (forward reads), müssen zu
einer Datei zusammengefügt werden, anschließend die auf _2 endenden (reverse
reads) zu einer zweiten Datei.
Wie fügt man nun mehrere Dateien zu einer zusammen?
Wer sich technisch fit genug fühlt, um die Windows-Eingabeaufforderung zu bedienen, kann das beispielsweise über den copy-Befehl erreichen:
copy /B “1_1.fq.gz“ + “2_1.fq.gz“ + “3_1.fq.gz“ “1.fq.gz“
copy /B “1_2.fq.gz“ + “2_2.fq.gz“ + “3_2.fq.gz“ “2.fq.gz“
(Das nur als Beispiel, falls man jeweils nur drei Dateien hätte. Tatsächlich sollten es mehr sein).
Wer sich nicht mit der Windows-Eingabeaufforderung auskennt, kann sich stattdessen mit Google ein einfach zu bedienendes Programm suchen, welches Dateien zusammenfügt. Theoretisch müsste es z.B. mit HJSplit gehen (dort auf den Button “Join” zum Zusammenfügen von Dateien klicken):
https://www.heise.de/download/product/hjsplit-55102
Ich persönlich habe HJSplit jedoch nicht ausprobiert, sondern es über die Windows-Eingabeaufforderung gelöst.
Fortan nur noch diese beiden FASTQ-Dateien verwenden, nicht die vielen anderen.
3.) Auf der linken Seite (“Tools”) nun “Get Data” anklicken, dann runterscrollen zu “Upload file from your computer”.
Hier die beiden FASTQ-Dateien hochladen. Wer keinen schnellen Internetzugang hat, wird leider lange Zeit warten müssen, bis alle Uploads abgeschlossen sind.
4.) Abwarten, bis beide Dateien fertig hochgeladen sind.
Auf der linken Seite (“Tools”) runterscrollen bis “Mapping”, draufklicken, dann auf “Map with BWA-MEM” klicken.
5.) Auf der nächsten Seite das Referenzgenom “hg38” auswählen (oder hg19 – je nachdem, was einem fehlt), “Select first set of reads” –> erste FASTQ-Datei, “Select second set of reads” –> zweite FASTQ-Datei, am Rest nichts ändern, “execute” klicken.
6.) 1-2 Tage später sollte die BAM-Datei fertig sein (sichtbar nach dem Login), so dass man sie sich theoretisch auf die eigene Festplatte herunterladen kann (dazu rechts bei “History” die neue BAM anklicken, dann auf das Disketten-Piktogramm klicken).
Falls man die Datei bei einem anderen Server einreichen möchte (z.B. YFull), dann ist es jedoch einfacher, einen Usegalaxy-Link zu generieren, den man einfach nur weitergibt. Diese Möglichkeit folgt in den Schritten 7-9:
7.) Einloggen. Auf der rechten Seite ist neben dem “History”-Schriftzug ein Zahnrad abgebildet. Da draufklicken.
8.) “Share or Publish” auswählen, dann “Make history accesible via link” anklicken.
NICHT die Option “[…] and publish” zusätzlich anklicken, sonst können alle anderen Benutzer von usegalaxy die eigenen Dateien sehen und auf diese zugreifen.
Schließlich die Option “Also make objects within the History accessible” anklicken.
9.) Auf der rechten Seite (“History”) die neu generierte BAM-Datei anklicken, dann das Disketten-Piktogramm anklicken, dann Rechtsklick auf “Download dataset” und den Link in die Zwischenablage kopieren. Dieser Link wird u.a. von YFull akzeptiert.
Ein paar Tage später, wenn YFull per Mail mitteilt, dass sie sich erfolgreich die Datei herunterladen konnten, ggf. wieder bei usegalaxy.eu einloggen, auf das Zahnrad klicken, und den Share-Link wieder abschalten.
Pingback: YFull – Erste Schritte – ΑΝΤΩΝΙΟΣ ΔΝΑ PROJECT